
File size: 8,913 Bytes
aec320b 87e8419 aec320b 02d3172 5604714 aec320b 87e8419 aec320b 4dc9bea aec320b 5604714 87e8419 aec320b 5604714 aec320b 5604714 aec320b 5604714 aec320b 5604714 aec320b 5604714 aec320b 02d3172 aec320b 5604714 aec320b 02d3172 aec320b 5604714 aec320b 5604714 aec320b 5604714 aec320b 5604714 aec320b 5604714 aec320b 5604714 aec320b 02d3172 aec320b 5604714 aec320b 5604714 aec320b |
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 72 73 74 75 76 77 78 79 80 81 82 83 84 85 86 87 88 89 90 91 92 93 94 95 96 97 98 99 100 101 102 103 104 105 106 107 108 109 110 111 112 113 114 115 116 117 118 119 120 121 122 123 124 125 126 127 128 129 130 131 132 133 134 135 136 137 138 139 140 141 142 143 144 145 146 147 148 149 150 151 152 153 154 155 156 157 158 159 160 161 162 163 164 165 166 167 168 169 170 171 172 173 174 175 176 177 178 179 180 181 182 183 184 185 186 187 188 189 190 191 192 193 194 195 196 |
---
base_model: ibm/biomed.sm.mv-te-84m
library_name: SmallMoleculeMultiView
license: apache-2.0
tags:
- chemistry
- model_hub_mixin
- molecules
- multiview
- pytorch
- pytorch_model_hub_mixin
---
# ibm/biomed.sm.mv-te-84m-MoleculeNet-ligand_scaffold-TOX21-101
`biomed.sm.mv-te-84m` is a biomedical foundation model for small molecules created using **MMELON** (**M**ulti-view **M**olecular **E**mbedding with **L**ate Fusi**on**), a flexible approach to aggregate multiple views (sequence, image, graph) of molecules in a foundation model setting. While models based on single view representation typically performs well on some downstream tasks and not others, the multi-view model performs robustly across a wide range of property prediction tasks encompassing ligand-protein binding, molecular solubility, metabolism and toxicity. It has been applied to screen compounds against a large (> 100 targets) set of G Protein-Coupled receptors (GPCRs) to identify strong binders for 33 targets related to Alzheimer’s disease, which are validated through structure-based modeling and identification of key binding motifs [Multi-view biomedical foundation models for molecule-target and property prediction](https://arxiv.org/abs/2410.19704).
- **Developers:** IBM Research
- **GitHub Repository:** [https://github.com/BiomedSciAI/biomed-multi-view](https://github.com/BiomedSciAI/biomed-multi-view)
- **Paper:** [Multi-view biomedical foundation models for molecule-target and property prediction](https://arxiv.org/abs/2410.19704)
- **Release Date**: Oct 28th, 2024
- **License:** [Apache 2.0](https://www.apache.org/licenses/LICENSE-2.0)
## Model Description
Source code for the model and finetuning is made available in [this repository](https://github.com/BiomedSciAI/biomed-multi-view).
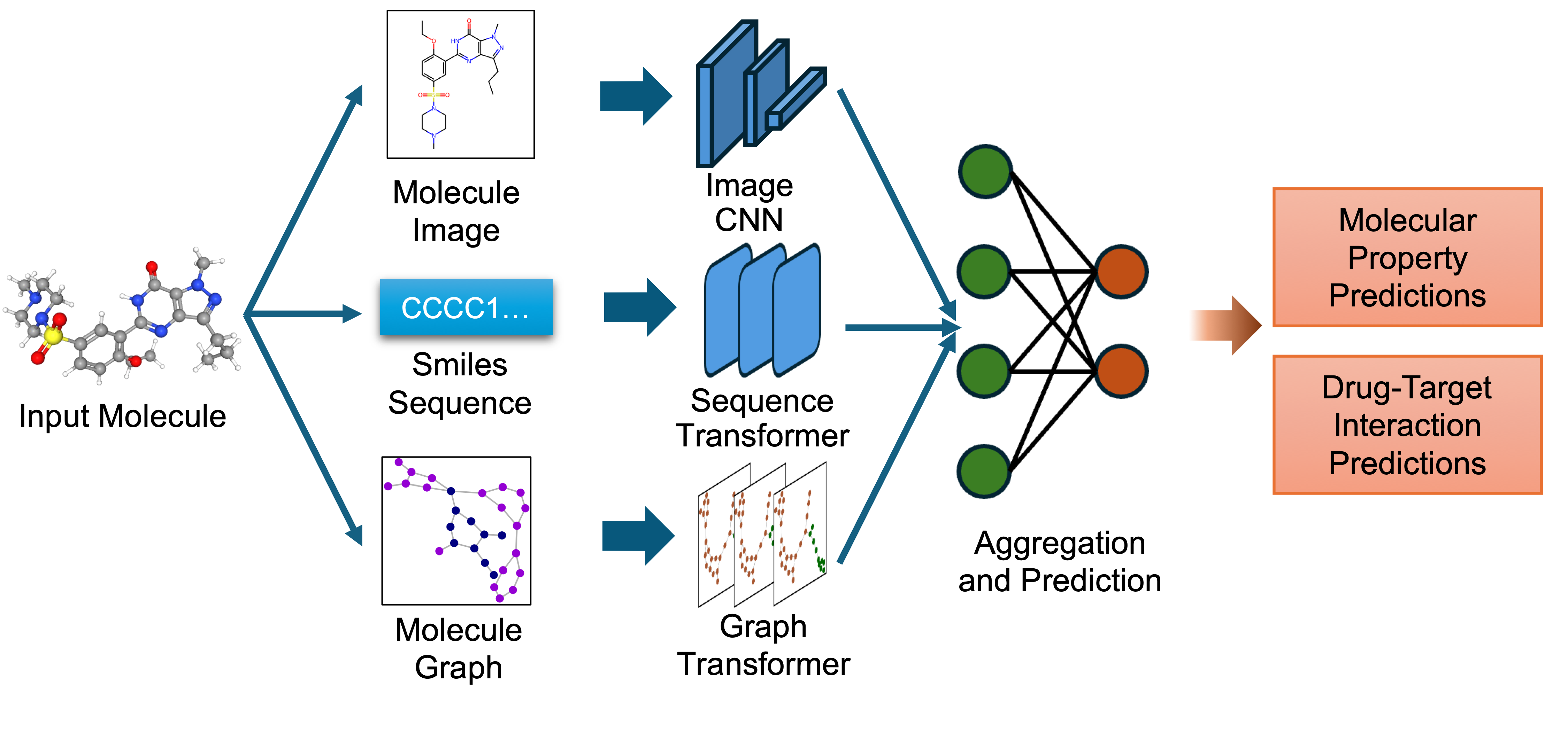
* Image Representation: Captures the 2D visual depiction of molecular structures, highlighting features like symmetry, bond angles, and functional groups. Molecular images are generated using RDKit and undergo data augmentation during training to enhance robustness.
* Graph Representation: Encodes molecules as undirected graphs where nodes represent atoms and edges represent bonds. Atom-specific properties (e.g., atomic number, chirality) and bond-specific properties (e.g., bond type, stereochemistry) are embedded using categorical embedding techniques.
* Text Representation: Utilizes SMILES strings to represent chemical structures, tokenized with a custom tokenizer. The sequences are embedded using a transformer-based architecture to capture the sequential nature of the chemical information.
The embeddings from these single-view pre-trained encoders are combined using an attention-based aggregator module. This module learns to weight each view appropriately, producing a unified multi-view embedding. This approach leverages the strengths of each representation to improve performance on downstream predictive tasks.
## Intended Use and Limitations
The model is intended for (1) Molecular property prediction. The pre-trained model may be fine-tuned for both regression and classification tasks. Examples include but are not limited to binding affinity, solubility and toxicity. (2) Pre-trained model embeddings may be used as the basis for similarity measures to search a chemical library. (3) Small molecule embeddings provided by the model may be combined with protein embeddings to fine-tune on tasks that utilize both small molecule and protein representation. (4) Select task-specific fine-tuned models are given as examples. Through listed activities, model may aid in aspects of the molecular discovery such as lead finding or optimization.
The model’s domain of applicability is small, drug-like molecules. It is intended for use with molecules less than 1000 Da molecular weight. The MMELON approach itself may be extended to include proteins and other macromolecules but does not at present provide embeddings for such entities. The model is at present not intended for molecular generation. Molecules must be given as a valid SMILES string that represents a valid chemically bonded graph. Invalid inputs will impact performance or lead to error.
## Usage
Using `SmallMoleculeMultiView` API requires the codebase [https://github.com/BiomedSciAI/biomed-multi-view](https://github.com/BiomedSciAI/biomed-multi-view)
## Installation
Follow these steps to set up the `biomed-multi-view` codebase on your system.
### Prerequisites
* Operating System: Linux or macOS
* Python Version: Python 3.11
* Conda: Anaconda or Miniconda installed
* Git: Version control to clone the repository
### Step 1: Set up the project directory
Choose a root directory where you want to install `biomed-multi-view`. For example:
```bash
export ROOT_DIR=~/biomed-multiview
mkdir -p $ROOT_DIR
```
#### Step 2: Create and activate a Conda environment
```bash
conda create -y python=3.11 --prefix $ROOT_DIR/envs/biomed-multiview
```
Activate the environment:
```bash
conda activate $ROOT_DIR/envs/biomed-multiview
```
#### Step 3: Clone the repository
Navigate to the project directory and clone the repository:
```bash
mkdir -p $ROOT_DIR/code
cd $ROOT_DIR/code
# Clone the repository using HTTPS
git clone https://github.com/BiomedSciAI/biomed-multi-view.git
# Navigate into the cloned repository
cd biomed-multi-view
```
Note: If you prefer using SSH, ensure that your SSH keys are set up with GitHub and use the following command:
```bash
git clone [email protected]:BiomedSciAI/biomed-multi-view.git
```
#### Step 4: Install package dependencies
Install the package in editable mode along with development dependencies:
``` bash
pip install -e .['dev']
```
Install additional requirements:
``` bash
pip install -r requirements.txt
```
#### Step 5: macOS-Specific instructions (Apple Silicon)
If you are using a Mac with Apple Silicon (M1/M2/M3) and the zsh shell, you may need to disable globbing for the installation command:
``` bash
noglob pip install -e .[dev]
```
Install macOS-specific requirements optimized for Apple’s Metal Performance Shaders (MPS):
```bash
pip install -r requirements-mps.txt
```
#### Step 6: Installation verification (optional)
Verify that the installation was successful by running unit tests
```bash
python -m unittest bmfm_sm.tests.all_tests
```
### Get embedding example
You can generate embeddings for a given molecule using the pretrained model with the following code.
```python
# Necessary imports
from bmfm_sm.api.smmv_api import SmallMoleculeMultiViewModel
from bmfm_sm.core.data_modules.namespace import LateFusionStrategy
# Load Model
model = SmallMoleculeMultiViewModel.from_pretrained(
LateFusionStrategy.ATTENTIONAL,
model_path="ibm/biomed.sm.mv-te-84m",
huggingface=True
)
# Load Model and get embeddings for a molecule
example_smiles = "CC(C)CC1=CC=C(C=C1)C(C)C(=O)O"
example_emb = SmallMoleculeMultiViewModel.get_embeddings(
smiles=example_smiles,
model_path="ibm/biomed.sm.mv-te-84m",
huggingface=True,
)
print(example_emb.shape)
```
### Get prediction example
You can use the finetuned models to make predictions on new data.
``` python
from bmfm_sm.api.smmv_api import SmallMoleculeMultiViewModel
from bmfm_sm.api.dataset_registry import DatasetRegistry
# Initialize the dataset registry
dataset_registry = DatasetRegistry()
# Example SMILES string
example_smiles = "CC(C)C1CCC(C)CC1O"
# Get dataset information for dataset
ds = dataset_registry.get_dataset_info("TOX21")
# Load the finetuned model for the dataset
finetuned_model_ds = SmallMoleculeMultiViewModel.from_finetuned(
ds,
model_path="ibm/biomed.sm.mv-te-84m-MoleculeNet-ligand_scaffold-TOX21-101",
inference_mode=True,
huggingface=True
)
# Get predictions
prediction = SmallMoleculeMultiViewModel.get_predictions(
example_smiles, ds, finetuned_model=finetuned_model_ds
)
print("Prediction:", prediction)
```
For more advanced usage, see our detailed examples at: https://github.com/BiomedSciAI/biomed-multi-view
## Citation
If you found our work useful, please consider giving a star to the repo and cite our paper:
```
@misc{suryanarayanan2024multiviewbiomedicalfoundationmodels,
title={Multi-view biomedical foundation models for molecule-target and property prediction},
author={Parthasarathy Suryanarayanan and Yunguang Qiu and Shreyans Sethi and Diwakar Mahajan and Hongyang Li and Yuxin Yang and Elif Eyigoz and Aldo Guzman Saenz and Daniel E. Platt and Timothy H. Rumbell and Kenney Ng and Sanjoy Dey and Myson Burch and Bum Chul Kwon and Pablo Meyer and Feixiong Cheng and Jianying Hu and Joseph A. Morrone},
year={2024},
eprint={2410.19704},
archivePrefix={arXiv},
primaryClass={q-bio.BM},
url={https://arxiv.org/abs/2410.19704},
}
``` |